A biopharma team runs the same plasma panel across three sites for a Phase II CNS trial. The molecule looks like it’s working at Site A. Site B shows a flat line. Site C’s data is unreadable — CVs north of 40%. Nobody changed the assay. Nobody changed the drug. What changed was everything upstream of the plate: the tube, the spin, the freezer, the isolation step. By the time the biomarker team notices, three months of samples are already compromised.
This is the reproducibility problem that rarely makes it into a grant proposal or a vendor slide deck, but it is one of the biggest reasons plasma-based biomarker data gets challenged in regulatory review and rejected in peer review. Sensitivity gets the marketing budget. Reproducibility is what actually determines whether a biomarker survives contact with a multi-site trial.
Key Takeaways
- Most plasma biomarker variability is introduced before the assay ever runs — at collection, processing, and storage. High-speed centrifugation alone can cut detectable EV-associated protein counts to roughly 30% of an unprocessed sample [1].
- Isolation-dependent methods add a second, compounding layer of variance: a six-laboratory comparison found inter-lab CVs on isolated vesicle concentration reaching 52.5% [2].
- Precision has three distinct levels — repeatability, intermediate precision, and inter-site (or inter-instrument) reproducibility — and most published EV biomarker studies report only the first [3].
- The ISEV Blood Task Force’s MIBlood-EV framework exists specifically because pre-analytical reporting has been inconsistent enough to undermine cross-study comparison [4].
- Well-controlled, isolation-free profiling approaches can bring assay precision into single-digit-to-low-teens CV ranges — but only when pre-analytical handling is standardized alongside the assay itself.
Where the Noise Enters: Pre-Analytical Variability
Every plasma biomarker study inherits variability from decisions made hours before a sample reaches the bench. Collection tube chemistry, time-to-processing, centrifugation speed, freeze-thaw history, and long-term storage conditions all measurably shift the composition of what ends up in the analysis. A 2026 study systematically testing these variables on plasma extracellular vesicle proteomes found that high-speed centrifugation (8,000 g) reduced detectable protein counts to roughly 30% of an unprocessed sample, rendering numerous established EV markers undetectable altogether [1]. The same study found that limited freeze-thaw cycling within a week had comparatively little effect on overall proteome coverage, while years of frozen storage progressively degraded both EV counts and proteome coverage — samples stored 20 years showed protein counts nearly as depleted as freshly high-speed-centrifuged plasma [1]. In other words, the assay itself was not the problem. The handling was, and different handling steps fail on very different timescales.
This is not a new observation, but it is chronically underweighted. Earlier work on pre-analytical variability in mid- and high-abundance plasma protein biomarkers identified the same handful of culprits — collection tube type, elapsed time before processing, and platelet activation during centrifugation — as dominant sources of measurement drift, well before EV-specific biomarkers were part of the conversation. That study found that mid- and high-abundance proteins were largely stable in unprocessed plasma left at room temperature for up to six hours, but that centrifugation temperature and tube choice measurably shifted cytokine concentrations [5]. The mechanisms carry over directly: plasma is a biologically active fluid from the moment it leaves the vein, and every additional hour, spin, or freeze cycle is an opportunity for the signal to shift.
For CNS biomarker programs specifically, this matters more than it does in most other indications. Multi-site Alzheimer’s, Parkinson’s, and ALS trials routinely span dozens of collection sites with variable phlebotomy training, different centrifuge models, and inconsistent freezer logistics. A biomarker platform that has excellent analytical sensitivity in a single, controlled lab can still fail in the field if it has no tolerance for that operational variability.

Isolation Adds Its Own Variance
Pre-analytical handling is only the first layer. Many plasma biomarker workflows add a second variable source: the isolation step itself. Differential ultracentrifugation, size-exclusion chromatography, and precipitation-based methods each recover a different, method-dependent subpopulation of the target material, and that recovery is sensitive to operator technique, reagent lot, and even ambient temperature.
The scale of this problem has been documented directly. A standardized multi-laboratory comparison using tunable resistive pulse sensing found that CVs on the concentration of vesicles isolated from citrated plasma reached as high as 52.5% when the same samples were analyzed across six different laboratories [2]. That is not a sensitivity failure — the instruments could detect the material just fine. It is a reproducibility failure rooted in how the isolation step interacts with operator- and site-specific variables. A separate variation study of nanoparticle tracking analysis across two instruments and multiple operators found that concentration measurements carried substantially more inter-assay variability (10–18% CV using identical settings) than size measurements (1–6% CV) on the very same samples, underscoring that isolation- and instrument-dependent concentration estimates are consistently the least reproducible part of the workflow [6].
A separate study examining methodological and short-term biological variation in plasma-derived EV surface and cargo proteins reached a related conclusion: methodological repeatability was reasonably good for particle sizing (3.3% CV) and mass spectrometry (8.2% CV), but markedly worse for surface-marker immunoassay measurements (22.6% CV) — variation of a similar order to genuine biological variability between individuals, which is precisely the failure mode that can erase a true treatment effect in a clinical dataset [3]. When method noise and biological signal are the same order of magnitude, no amount of downstream statistical correction fully recovers the difference.
This is the core argument for isolation-free approaches to plasma biomarker profiling: every isolation step is a hand-off point where variance gets added, and removing that hand-off removes an entire category of possible failure — not just one source of noise, but a compounding one that interacts unpredictably with everything upstream.
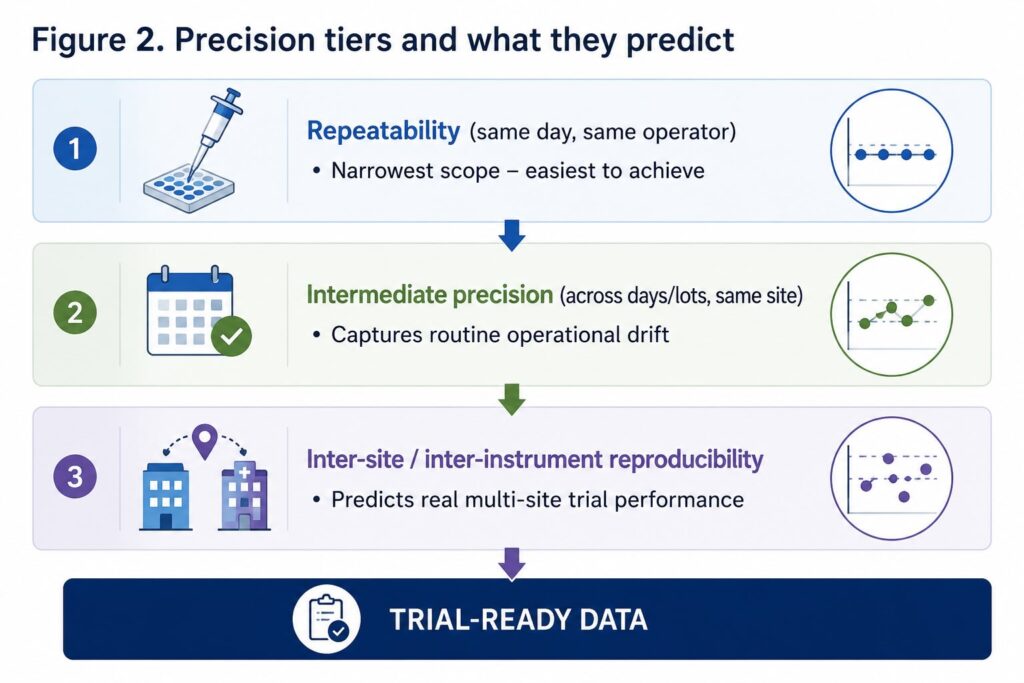
Defining What “Reproducible” Actually Means
Part of why this problem persists is a vocabulary gap. “Reproducible” is used loosely in biomarker literature to mean everything from “I got the same number twice” to “six independent labs agreed.” These are very different claims, and conflating them is how underpowered validation data ends up presented as robust.
Precision, properly defined, has three tiers:
- Repeatability (intra-assay precision): the variation between replicates run in the same assay, same day, same operator. In one instrument-variation study, intra-assay CVs for particle concentration ranged from 5–12% within a single day on a single instrument [6] — the easiest number to generate, and the least informative on its own.
- Intermediate precision: variation across different days, operators, or reagent lots within the same lab. The same study found inter-assay (day-to-day) CVs on concentration rising to 10–18% even on identically configured instruments [6].
- Inter-site/inter-instrument reproducibility: variation when the same samples are run at genuinely different sites or on different instruments. This is the number that predicts what will actually happen in a multi-site Phase II trial — and it is the number most likely to reveal problems that single-lab repeatability data conceals.
The ISEV Blood Task Force’s MIBlood-EV framework was developed specifically to address this gap, proposing standardized reporting of blood collection, processing, and quality-control variables so that reproducibility claims across studies can actually be compared on equal footing [4]. It builds on the broader MISEV consensus guidelines for EV research reporting more generally [7]. Any plasma biomarker platform — including EV-based ones — should be evaluated against all three precision tiers, not just the first.
What Controlled Precision Looks Like
None of this is an argument against plasma biomarkers — it’s an argument for treating reproducibility as a primary design constraint rather than an afterthought reported in a supplementary table. Two design choices do most of the work: standardizing pre-analytical handling with the same rigor applied to the assay itself, and minimizing the number of hand-off steps between sample and readout.
This is the design philosophy behind LuminEV‘s approach to plasma biomarker profiling. Rather than requiring a separate isolation step before analysis, the platform enriches for neuron-derived signal directly within a multiplexed, bead-based immunoassay format — at the mechanism level, profiling surface-exposed proteins on circulating vesicles without an upstream separation step that would otherwise introduce its own operator- and site-dependent variance. In current validation work, this isolation-free format has demonstrated assay precision with CV below 15%, a range that compares favorably with the isolation-dependent inter-lab variability documented above [2, 6]. That single design decision — removing the isolation hand-off rather than trying to standardize it — is what helps close the gap between single-site pilot data and multi-site trial-grade reproducibility.
The point isn’t that isolation-free is a universal fix; it’s that reproducibility has to be engineered into the workflow at the same stage decisions get made about sensitivity and specificity, not audited afterward.
Building a Reproducibility-First Workflow: What to Do Next
For teams evaluating or building a plasma biomarker program for a CNS trial, a few concrete steps reduce risk before the first sample is ever run:
- Standardize and document pre-analytical variables (tube type, time-to-spin, storage duration) using a framework like MIBlood-EV [4], even if the assay itself isn’t EV-specific.
- Ask any assay vendor for inter-site or inter-instrument reproducibility data, not just intra-assay CV — and be specific about which precision tier is being reported.
- Where possible, favor workflows that minimize manual hand-off steps between sample collection and readout, since each step is an independent source of compounding variance.
- Pilot the assay across at least two operators or sites before committing to a full trial protocol, even if timeline pressure argues against it.
- Treat high-speed centrifugation and long-term frozen storage as the highest-risk points in a biobank-dependent study design, given how sharply they can deplete EV-associated signal [1].
The field is moving toward standardized pre-analytical reporting as a baseline expectation rather than a nice-to-have, and reproducibility data is increasingly requested during protocol review rather than after data lock. Plasma biomarker platforms that can show inter-site precision — not just single-lab sensitivity — will be the ones that survive that scrutiny.
References
[1] Suresh PS, Zhang Q. Impact of preanalytical factors on plasma extracellular vesicles and human plasma proteome. Clin Proteom. 2026;23(1):15. https://doi.org/10.1186/s12014-026-09590-8
[2] Vogel R, Coumans FAW, Maltesen RG, et al. A standardized method to determine the concentration of extracellular vesicles using tunable resistive pulse sensing. J Extracell Vesicles. 2016;5:31242. https://doi.org/10.3402/jev.v5.31242
[3] Krzyslak H, Szejniuk WM, Falkmer U, Honoré B, Jørgensen MM, Sten C, Pedersen S, Christiansen G, Kristensen SR. Methodological and Short-Term Diurnal Variation in Surface and Cargo Proteins in Plasma Extracellular Vesicles. Curr Issues Mol Biol. 2026;48:120. https://doi.org/10.3390/cimb48010120
[4] Lucien F, Gustafson D, Lenassi M, Li B, Teske JJ, Boilard E, et al. MIBlood-EV: Minimal information to enhance the quality and reproducibility of blood extracellular vesicle research. J Extracell Vesicles. 2023;12(12):e12385. https://doi.org/10.1002/jev2.12385
[5] Aguilar-Mahecha A, Kuzyk MA, Domanski D, Borchers CH, Basik M. The Effect of Pre-Analytical Variability on the Measurement of MRM-MS-Based Mid- to High-Abundance Plasma Protein Biomarkers and a Panel of Cytokines. PLoS ONE. 2012;7(6):e38290. https://doi.org/10.1371/journal.pone.0038290
[6] Vestad B, Llorente A, Neurauter A, Phuyal S, Kierulf B, Kierulf P, Skotland T, Sandvig K, Haug KBF, Øvstebø R. Size and concentration analyses of extracellular vesicles by nanoparticle tracking analysis: a variation study. J Extracell Vesicles. 2017;6(1):1344087. https://doi.org/10.1080/20013078.2017.1344087
[7] Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. https://doi.org/10.1080/20013078.2018.1535750
All seven references above have been verified against their original source pages (PMC, journal publisher sites, or DOI resolution) — author lists, journal names, volumes, and years are confirmed as of this draft.