
Blood biomarkers for neurological disease have a fundamental problem: the brain is remote. Here is why neuron-derived EVs are changing what researchers can measure — and how a multiplexed panel spans ALS, Parkinson’s, and Alzheimer’s research in a single workflow.
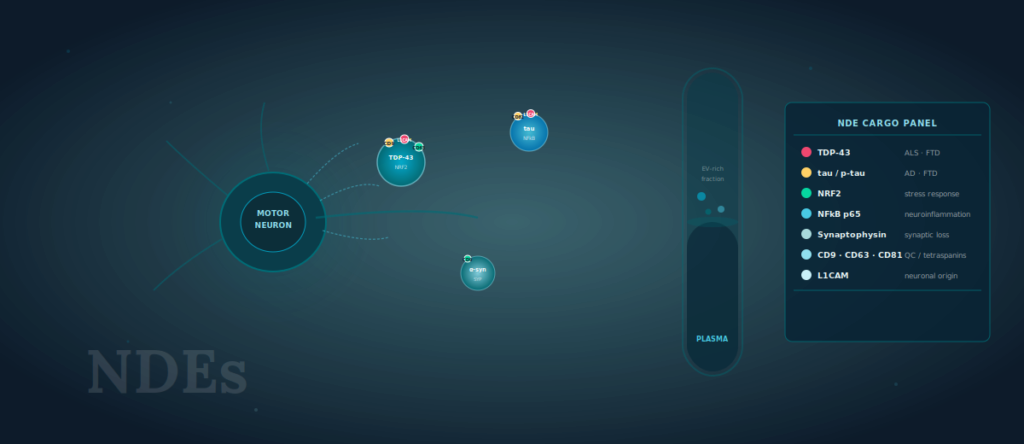
Neuron-derived extracellular vesicles (NDEs) carry disease-relevant intracellular cargo — including TDP-43, tau, NRF2, NFkB, and synaptic proteins — from the central nervous system into peripheral blood, enabling minimally invasive biomarker access across ALS, Parkinson’s disease, and Alzheimer’s disease research programs.
KEY TAKEAWAYS
- NfL and GFAP reflect neuronal damage but lack the specificity to distinguish disease mechanisms or cell types.
- Neuron-derived EVs carry intracellular cargo — TDP-43, tau, NRF2, NFkB, synaptic proteins — directly from neurons into peripheral blood.
- A multiplexed NDE surface protein panel can serve ALS, PD, and AD biomarker programs within a single standardized workflow.
- Tetraspanins (CD9, CD63, CD81) function as both quality controls and phenotyping anchors when profiling neuron-derived subpopulations.
- Pre-analytical handling, sample volume, and assay throughput are the three variables most likely to determine whether a plasma EV study succeeds at cohort scale
Why NfL and GFAP Are Not Enough
Neurofilament light chain has become the most widely used blood biomarker in neurological clinical trials, and for good reason. Plasma NfL concentrations correlate with rates of neurodegeneration across ALS, multiple sclerosis, frontotemporal dementia, and Alzheimer’s disease, and the assay is sensitive enough to detect longitudinal changes within individual patients [1]. GFAP, a marker of astrocytic injury, adds complementary information about glial involvement [2].
But both markers have a ceiling. NfL reflects the rate of axonal damage — it does not distinguish whether that damage is driven by TDP-43 aggregation, tau pathology, alpha-synuclein propagation, or mitochondrial failure. In a Phase II trial where the drug target is TDP-43 mislocalization, a reduction in NfL tells you neurons are dying more slowly. It does not tell you whether TDP-43 nuclear clearance is recovering [3]. For mechanistic trials, that gap is significant.
GFAP has similar limitations. Elevated GFAP is a downstream consequence of neuroinflammation; it doesn’t resolve which inflammatory pathway — NFkB-driven cytokine release, NRF2 stress response, complement activation — is dominant in a given patient or cohort. The result is a common frustration in translational neuroscience: highly sensitive assays measuring the wrong readout for the mechanistic question being asked.
What Neuron-Derived EVs Carry That Plasma Cannot
Extracellular vesicles are membrane-bound particles, typically 30–200nm in diameter, released by virtually all cell types as part of normal physiology and in response to cellular stress [4]. The cargo they carry — proteins, nucleic acids, lipids — reflects the state of the cell of origin at the time of release. An EV released by a stressed motor neuron carries a molecular record of that stress into the circulation.
The practical challenge has been specificity. Plasma is a mixture of EVs from hundreds of cell types: platelets, red blood cells, endothelial cells, hepatocytes, immune cells. Measuring total EV cargo in unfractionated plasma conflates signals from all of them. To use EVs as neuron-specific reporters, you need a method for enriching or identifying the neuron-derived subpopulation.
L1CAM (CD171), a neural cell adhesion molecule expressed on the surface of neuron-derived EVs, has been used as an immunocapture target to enrich NDEs from plasma [5]. Studies using L1CAM-based enrichment have detected TDP-43, tau, alpha-synuclein, NRF2, NFkB p65, and synaptic proteins including synaptophysin in plasma NDEs from patients with ALS, AD, and PD at concentrations distinguishable from healthy controls [5, 6, 7]. What this means in practice: cargo that is entirely intracellular — TDP-43 normally resides in the nucleus and does not freely circulate in plasma — becomes measurable in peripheral blood through the EV compartment. That is a qualitatively different kind of biomarker access.
Disease-Specific Applications: ALS, Parkinson’s, and Alzheimer’s
The same EV profiling workflow maps onto different mechanistic questions depending on the disease context. This is one of the underappreciated practical advantages of a multiplexed panel: a single standardized assay generates disease-relevant readouts across research programs without requiring separate assay development for each indication.
ALS and Frontotemporal Dementia
TDP-43 mislocalization — from the nucleus into cytoplasmic aggregates — is the defining pathological event in approximately 97% of ALS cases and around 45% of FTD cases [8]. In plasma NDEs, TDP-43 cargo reflects this mislocalization: studies have shown that TDP-43 levels in L1CAM-enriched EVs are elevated in ALS patients compared to healthy controls and correlate with disease progression as measured by ALSFRS-R [6].
NFkB p65 and NRF2 add a neuroinflammatory dimension. NFkB is a master regulator of pro-inflammatory cytokine transcription; NRF2 governs the antioxidant stress response. In ALS, the balance between these pathways in motor neurons reflects whether the cellular environment is predominantly inflammatory or attempting adaptive stress compensation — a distinction with direct relevance to clinical trials targeting neuroinflammation [3]. Synaptic markers — synaptophysin, GAP-43 — provide a third layer: evidence of synaptic terminal loss that may precede axonal degeneration as measured by NfL [9].
Parkinson’s Disease
Alpha-synuclein aggregation and spreading is the central pathological mechanism in PD, and plasma NDE levels of alpha-synuclein have been studied as a potential staging biomarker [7]. Beyond alpha-synuclein, the same oxidative stress framework relevant in ALS applies in PD, where complex I inhibition and mitochondrial fragmentation are well-established upstream events [10]. NRF2 activation as a compensatory response to mitochondrial dysfunction is directly measurable in the NDE compartment.
One practical advantage in PD research is sample availability: large longitudinal cohorts with long prodromal phases — PPMI, UK Biobank subsets — make it feasible to study EV biomarker trajectories years before motor symptom onset. A multiplexed EV panel run on archived plasma from these cohorts could identify which cargo patterns predict conversion, a study design that single-marker assays make logistically difficult due to per-marker cost and sample volume constraints.
Alzheimer’s Disease
Tau is the most extensively studied NDE cargo marker in AD. Phosphorylated tau species in plasma have shown strong diagnostic performance for amyloid/tau pathology [2], but NDE-associated tau provides an additional dimension: it reflects tau that has been packaged and released by neurons, potentially representing an early stage of trans-synaptic tau propagation [11].
Studies using L1CAM-enriched NDEs have shown that cargo ratios — the ratio of phospho-tau to total tau, or NRF2 to NFkB — outperform single-marker concentrations in distinguishing AD subtypes and tracking cognitive decline [5]. This multiplexing advantage is not achievable with single-plex assay formats, and represents one of the strongest arguments for moving to a panel-based approach in AD biomarker research.
Surface Markers as Quality Controls and Phenotyping Anchors
Any discussion of NDE cargo measurement has to address a methodological reality: the quality of the EV preparation determines the reliability of the cargo readout. Tetraspanins — CD9, CD63, and CD81 — are the established surface markers for EV characterization per MISEV2023 guidelines, and their measurement serves two functions in NDE biomarker studies [4].
First, tetraspanin ratios provide a within-sample quality control. A plasma preparation with normal total EV yield but anomalous CD9:CD63 ratios may indicate platelet activation during collection, red blood cell contamination, or pre-analytical handling issues — all of which confound cargo measurements. Tracking tetraspanins across a cohort flags outlier samples before they propagate into downstream analysis.
Second, tetraspanin profiling contributes to NDE phenotyping. L1CAM-enriched EVs from neuronal sources show distinct surface protein patterns compared to EVs of glial or endothelial origin. Including tetraspanins alongside neuronal surface markers in the panel adds confidence that the enriched population is genuinely neuron-derived rather than contaminated with EVs from peripheral cells expressing L1CAM at low levels [4, 12].
Designing a Plasma EV Biomarker Study That Scales
The scientific rationale for NDE profiling is well established. The practical challenges sit in study design — and three variables determine whether a plasma EV biomarker study generates usable data or becomes a source of irreproducibility.
Sample volume and collection protocol. Most NDE enrichment workflows require 0.5–2 mL of plasma. Pre-analytical handling is the key constraint: platelet depletion spin speed, time to processing, freeze-thaw cycles, and anticoagulant choice (EDTA vs. citrate) all affect EV yield and cargo integrity [4, 13]. Establishing a standardized operating procedure before the first sample is collected — not after — is the single most impactful thing a study team can do for data quality.
Throughput and cohort size. A biomarker discovery study in ALS typically requires 50–100 patients plus matched controls to achieve adequate statistical power for identifying markers that track ALSFRS-R progression. An assay format that processes one sample at a time creates a bottleneck that compounds across a 150-sample cohort. Transitioning to a 96-well multiplexed format — where 8 or more targets are measured simultaneously per sample — compresses assay time and reduces inter-run variability [14].
Multiplexing vs. single-plex tradeoffs. Running TDP-43, NRF2, NFkB, and three tetraspanins as separate assays from the same plasma aliquot introduces sample splitting, additional freeze-thaw events, and inter-assay normalization challenges. A single multiplexed assay run from one aliquot eliminates these variables and preserves sample for downstream validation. For cohorts where samples are irreplaceable — longitudinal ALS biobank collections, for instance — this is not a minor operational consideration.
The LuminEV Research Kit was developed specifically for this workflow: multiplexed EV surface protein measurement from plasma, in a 96-well format, with pre-validated panels covering neuronal and tetraspanin markers. For teams designing studies in ALS, PD, or AD where sample conservation and throughput are real constraints, it addresses the gap between the scientific rationale for NDE profiling and the operational reality of running it at cohort scale.

What the Field Is Moving Toward
The next phase of NDE biomarker research is moving in two directions simultaneously. On the discovery side, single-vesicle analysis methods — including nano-flow cytometry and single-EV approaches — are beginning to resolve cargo heterogeneity within the NDE population, distinguishing vesicles that carry TDP-43 from those that do not within the same patient sample [15]. This sub-population resolution may prove important for understanding which neurons are most affected and at what disease stage.
On the clinical translation side, the ISEV community and regulatory agencies are actively working toward biomarker qualification frameworks for EV-based assays [4]. Several academic-pharma consortia are building the evidentiary packages needed for formal qualification — which means the data generated in exploratory NDE studies today may directly inform the assays used in late-phase trials within the decade. For researchers entering this space now, the methodological choices made at the study design phase will determine whether their datasets contribute to that effort or remain isolated findings. Standardization is not a constraint on innovation; it is the condition that makes NDE biomarker data comparable, reproducible, and ultimately useful in the clinic.
References
- Zetterberg H, Blennow K. Moving fluid biomarkers for Alzheimer’s disease from research tools to routine clinical diagnostics. Mol Neurodegener. 2021;16(1):10. doi:10.1186/s13024-021-00430-x
- Benedet AL, Milà-Alomà M, Vrillon A, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78(11):1325–1335. doi:10.1001/jamaneurol.2021.2713
- Goutman SA, Hardiman O, Al-Chalabi A, et al. Recent advances in the diagnosis and prognosis of amyotrophic lateral sclerosis. Lancet Neurol. 2022;21(5):480–493. doi:10.1016/S1474-4422(21)00465-X
- Welsh JA, Goberdhan DCI, O’Driscoll L, et al. Minimal information for studies of extracellular vesicles (MISEV2023). J Extracell Vesicles. 2024;13(2):e12451.
- Mustapic M, Eitan E, Werner JK Jr, et al. Plasma extracellular vesicles enriched for neuronal origin: a potential window into brain pathologic processes. Front Neurosci. 2017;11:278. doi:10.3389/fnins.2017.00278
- Benussi L, Fontana F, Ghidoni R, et al. TDP-43 in cerebrospinal fluid and extracellular vesicles: implications for ALS diagnosis. Cells. 2020;9(11):2471. doi:10.3390/cells9112471
- Goetzl EJ, Elahi FM, Merrill DA, et al. Altered plasma neuron-derived exosomes in frontotemporal dementia. Ann Clin Transl Neurol. 2019;6(9):1696–1702. doi:10.1002/acn3.50853
- Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi:10.1126/science.1134108
- Liu Y, Bhatt S, Wang Y, et al. Synaptic vesicle proteins and EV cargo reflect pre-symptomatic synaptic loss in ALS mouse models. Neurobiol Dis. 2022;168:105714.
- Poewe W, Seppi K, Tanner CM, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013. doi:10.1038/nrdp.2017.13
- Ding X, Ma M, Teng J, et al. Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes. Elife. 2015;4:e09525.
- Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018). J Extracell Vesicles. 2018;7(1):1535750. doi:10.1080/20013078.2018.1535750
- Lacroix R, Judicone C, Mooberry M, et al. Standardization of pre-analytical variables in plasma microparticle determination. J Thromb Haemost. 2013;11(6):1190–1193.
- Aguilar PP, Schneider TA, Ghorbanpour S, et al. Bead-based multiplex detection of EV surface proteins without prior isolation. J Extracell Vesicles. 2022;11(4):e12210.
- Koo CZ, Harrison P, Lannigan J, et al. Advances in single extracellular vesicle analysis. Lab Chip.2023;23(5):1253–1274. doi:10.1039/D2LC00898G

